About us
Learn how GA4GH helps expand responsible genomic data use to benefit human health.
Learn how GA4GH helps expand responsible genomic data use to benefit human health.
Our Strategic Road Map defines strategies, standards, and policy frameworks to support responsible global use of genomic and related health data.
Discover how a meeting of 50 leaders in genomics and medicine led to an alliance uniting more than 5,000 individuals and organisations to benefit human health.
GA4GH Inc. is a not-for-profit organisation that supports the global GA4GH community.
The GA4GH Council, consisting of the Executive Committee, Strategic Leadership Committee, and Product Steering Committee, guides our collaborative, globe-spanning alliance.
The Funders Forum brings together organisations that offer both financial support and strategic guidance.
Learn how GA4GH is working to expand its global reach and cultivate connection and meaningful engagement with members of the international genomics and health community.
Distributed across a number of Host Institutions, our staff team supports the mission and operations of GA4GH.
Curious who we are? Meet the people and organisations across six continents who make up GA4GH.
More than 500 organisations connected to genomics — in healthcare, research, patient advocacy, industry, and beyond — have signed onto the mission and vision of GA4GH as Organisational Members.
These core Organisational Members are genomic data initiatives that have committed resources to guide GA4GH work and pilot our products.
This subset of Organisational Members whose networks or infrastructure align with GA4GH priorities has made a long-term commitment to engaging with our community.
Local and national organisations assign experts to spend at least 30% of their time building GA4GH products.
Anyone working in genomics and related fields is invited to participate in our inclusive community by creating and using new products.
Wondering what GA4GH does? Learn how we find and overcome challenges to expanding responsible genomic data use for the benefit of human health.
Study Groups define needs. Participants survey the landscape of the genomics and health community and determine whether GA4GH can help.
Work Streams create products. Community members join together to develop technical standards, policy frameworks, and policy tools that overcome hurdles to international genomic data use.
GIF solves problems. Organisations in the forum pilot GA4GH products in real-world situations. Along the way, they troubleshoot products, suggest updates, and flag additional needs.
GIF Projects are community-led initiatives that put GA4GH products into practice in real-world scenarios.
The GIF AMA programme produces events and resources to address implementation questions and challenges.
NIF finds challenges and opportunities in genomics at a global scale. National programmes meet to share best practices, avoid incompatabilities, and help translate genomics into benefits for human health.
Communities of Interest find challenges and opportunities in areas such as rare disease, cancer, and infectious disease. Participants pinpoint real-world problems that would benefit from broad data use.
The Technical Alignment Subcommittee (TASC) supports harmonisation, interoperability, and technical alignment across GA4GH products.
Find out what’s happening with up to the minute meeting schedules for the GA4GH community.
See all our products — always free and open-source. Do you work on cloud genomics, data discovery, user access, data security or regulatory policy and ethics? Need to represent genomic, phenotypic, or clinical data? We’ve got a solution for you.
All GA4GH standards, frameworks, and tools follow the Product Development and Approval Process before being officially adopted.
Learn how other organisations have implemented GA4GH products to solve real-world problems.
Help us transform the future of genomic data use! See how GA4GH can benefit you — whether you’re using our products, writing our standards, subscribing to a newsletter, or more.
Join our community! Explore opportunities to participate in or lead GA4GH activities.
Help create new global standards and frameworks for responsible genomic data use.
Align your organisation with the GA4GH mission and vision.
Want to advance both your career and responsible genomic data sharing at the same time? See our open leadership opportunities.
Join our international team and help us advance genomic data use for the benefit of human health.
Discover current opportunities to engage with GA4GH. Share feedback on our products, apply for volunteer leadership roles, and contribute your expertise to shape the future of genomic data sharing.
Solve real problems by aligning your organisation with the world’s genomics standards. We offer software dvelopers both customisable and out-of-the-box solutions to help you get started.
Learn more about upcoming GA4GH events. See reports and recordings from our past events.
Speak directly to the global genomics and health community while supporting GA4GH strategy.
Be the first to hear about the latest GA4GH products, upcoming meetings, new initiatives, and more.
Questions? We would love to hear from you.
Read news, stories, and insights from the forefront of genomic and clinical data use.
Publishes regular briefs exploring laws and regulations, including data protection laws, that impact genomic and related health data sharing
Translates findings from studies on public attitudes towards genomic data sharing into short blog posts, with a particular focus on policy implications
Attend an upcoming GA4GH event, or view meeting reports from past events.
See new projects, updates, and calls for support from the Work Streams.
Read academic papers coauthored by GA4GH contributors.
Listen to our podcast OmicsXchange, featuring discussions from leaders in the world of genomics, health, and data sharing.
Check out our videos, then subscribe to our YouTube channel for more content.
View the latest GA4GH updates, Genomics and Health News, Implementation Notes, GDPR Briefs, and more.
1 Oct 2019
CRAM is an international community standard file format for storing compressed DNA sequencing data. In this post, adapted from an earlier post on his personal blog Data Geekdom, James Bonfield dispels a few myths about the format and its ability to meet the community’s needs for low cost storage of large scale sequencing data. Bonfield is a member of the GA4GH Large Scale Genomics Work Stream and a software developer at the Sanger Institute. He is the primary maintainer of the CRAM standard, which was originally theorized in 2010 in a paper by Fritz et al.

By James Bonfield
CRAM is an international community standard file format for storing compressed DNA sequencing data. In this post, adapted from an earlier post on his personal blog Data Geekdom, James Bonfield dispels a few myths about the format and its ability to meet the community’s needs for low cost storage of large scale sequencing data. Bonfield is a member of the GA4GH Large Scale Genomics Work Stream and a software developer at the Sanger Institute. He is the primary maintainer of the CRAM standard, which was originally theorized in 2010 in a paper by Fritz et al.
To kick things off, I want to remind you of the most important thing about CRAM: it is a file format. CRAM does not equal Samtools or Picard—it is a format, with several independent implementations, which include htslib for use on Samtools and htsjdk for use on Picard. Please, please, please do not confuse the format with its implementations.
At GA4GH we believe in the idea of collaborating on standards and competing on implementations. Our community is dedicated to maintaining viable, open standards that work for everyone. If an implementation has a flaw or weakness, its developers should put effort into improving it, rather than producing a whole new format.
As a human genetics researcher you may not always have a reference genome to compare against your sequence data—that shouldn’t prevent you from using an efficient file format. While, the main implementations of CRAM encode genomic sequence data as a difference from the reference genome, this is not mandatory. The current software packages for reading and writing CRAM files—which include both htslib and htsjdk—have modes for working without a reference.
Technically Speaking
CRAM supports three modes of operation with regards to references.
These can be enabled using options in Samtools. For example:

For embedded references our implementations do sadly have weaknesses. If we read a CRAM file using an embedded reference and wish to output a new modified CRAM also using an embedded reference, then somehow we need to get that embedded reference on disk so we can rewrite it again using “no_ref” mode. Currently this is hard to do. Ideally the tools would simply generate embedded references on the fly as the consensus of the aligned data within the CRAM file. This may even be more efficient than using an external reference in some scenarios.
If you’re working without a public reference or doing a de novo assembly, you might want to upload a file of unaligned data to ENA, NCBI, or another data repository in order to… CRAM can do this quite happily, and using much less disc space than a FASTQ file. If you’re thinking about the tools that need FASTQ to run, fear not as CRAM files can be converted back to FASTQ on the fly.
Technically Speaking
The samtools fastq command can turn a CRAM file back into FASTQ file(s). The Staden “io_lib” package for reading bioinformatics / DNA Sequence formats includes a CRAM filter tool that permits extraction of specific CRAM slices based on their numerical IDs if the file has been indexed. For example:

You can specify – or /dev/stdout for the output to permit streaming. When combined with samtools fastq this permits subsets of FASTQ to be generated and so works better in a map/reduce style manner than streaming the entire file and dispatching commands. Nevertheless, the tool chain is currently clunky and we are actively working to tidy this up.
Human genetics researchers need a reliably lossless file format—that is, you need to be confident that you won’t lose your data when you switch between formats. While the original Cramtools code had lossy compression enabled by default, the tools for reading and writing today’s CRAM files (eg., Samtools, Scramble, and Picard) are indeed lossless by default. That said, CRAM can still do lossy compression if desired.
Technically Speaking
Malformed data and a few very quirky corner cases aside, CRAM is lossless in that a round trip from BAM to CRAM to BAM will return the same data.
Technically there are a few legitimate things which CRAM cannot store, such as distinguishing between “=” / “X” CIGAR operators and “M” and knowledge of whether the read had an NM/MD tag or not (it’ll just regenerate these irrespective of whether they were in the original data)—anything else that does not survive the “round-trip” (e.g., CIGAR fields for unmapped data) is either broken data or information, which the SAM specification states have no meaning anyway. Indeed, CRAM losslessness is comparable to BAM, which, when converting from and back to SAM can result in changes such as uppercasing sequence and, for Picard, changing some auxiliary types (H to B).
In addition to its initial implementers, EMBL-EBI and Sanger, CRAM is now being used across the world’s leading bioinformatics institutions, including Genomics England, Australian Genomics, the Broad Institute of MIT and Harvard, Washington University at St. Louis.
More alignment files have been deposited in the EBI’s ENA and EGA archives in CRAM than BAM, by a factor of around 2:1—that’s well over a million CRAM files on one site alone. The Broad Institute also have wide adoption of CRAM, more focused on per-project than an overall strategy, but with more projects switching over as the space savings are so large.
The code base has been downloaded from conda over 1 million times, plus many more from github. Htslib has also been embedded into 100s of other github projects. Obviously, most of these downloads will not be specifically for CRAM, but if you use htslib you’ll have the ability to use CRAM. Similarly for htsjdk. Since many programming languages have interfaces to htslib and/or htsjdk, CRAM is also supported by many languages.
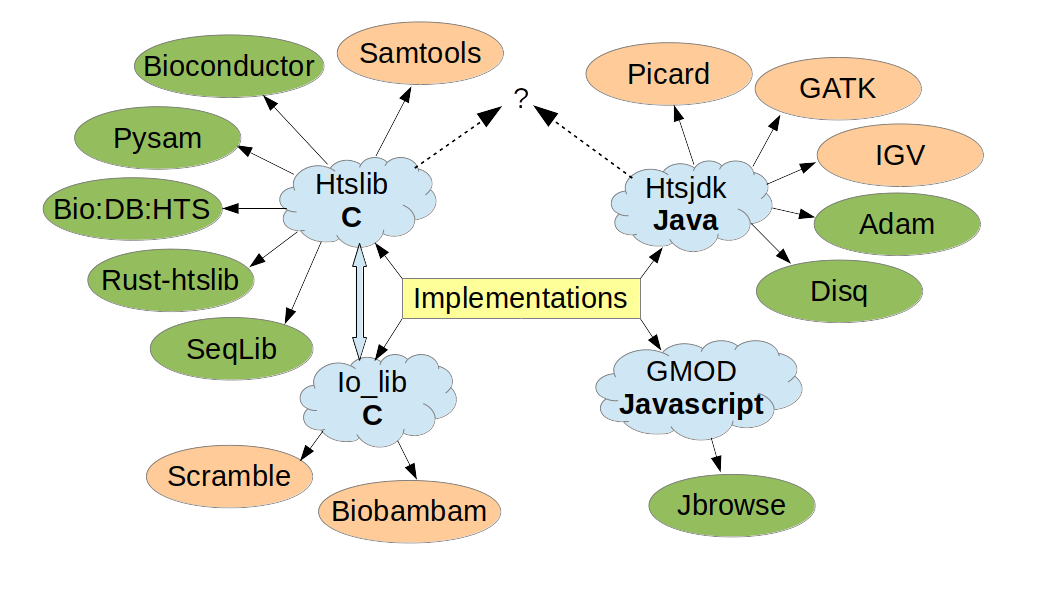 Figure 1: CRAM implementations
Figure 1: CRAM implementations
There are numerous comparisons of CRAM to alternative formats that gloss over a huge wealth of detail. CRAM files can differ in size by implementation, but even within the same implementation there are different levels of compression depending on whether the user is targeting speed, file size, or granularity of random access. Comparisons between CRAM and other tools must include not just size but also the software, version, and command line options being used.
Technically Speaking
Just like gzip, scramble has -1 to -9 arguments to favour light or heavy levels of compression. By default, neither Scramble nor Samtools uses the bzip2 and lzma codecs, but they can be enabled for higher compression. The number of sequences per slice also improves the compression ratio, albeit at the cost of less fine grained random access. For example:

It is also possible to reduce the number of sequences per slice (which defaults to 10,000) in order to permit finer-grained random access.
We are currently working on a prototype of CRAM 3.1 with more compression codecs. Performance on a set of NovaSeq alignments is demonstrated below showing the tradeoff between size vs speed (“normal” vs “archive” modes). For comparison, BAM speed is also included, but note the file size is a long way off the edge of this plot at 70Gb. The benchmarks on htslib.org offer further examples of how size and speed trade-offs work.
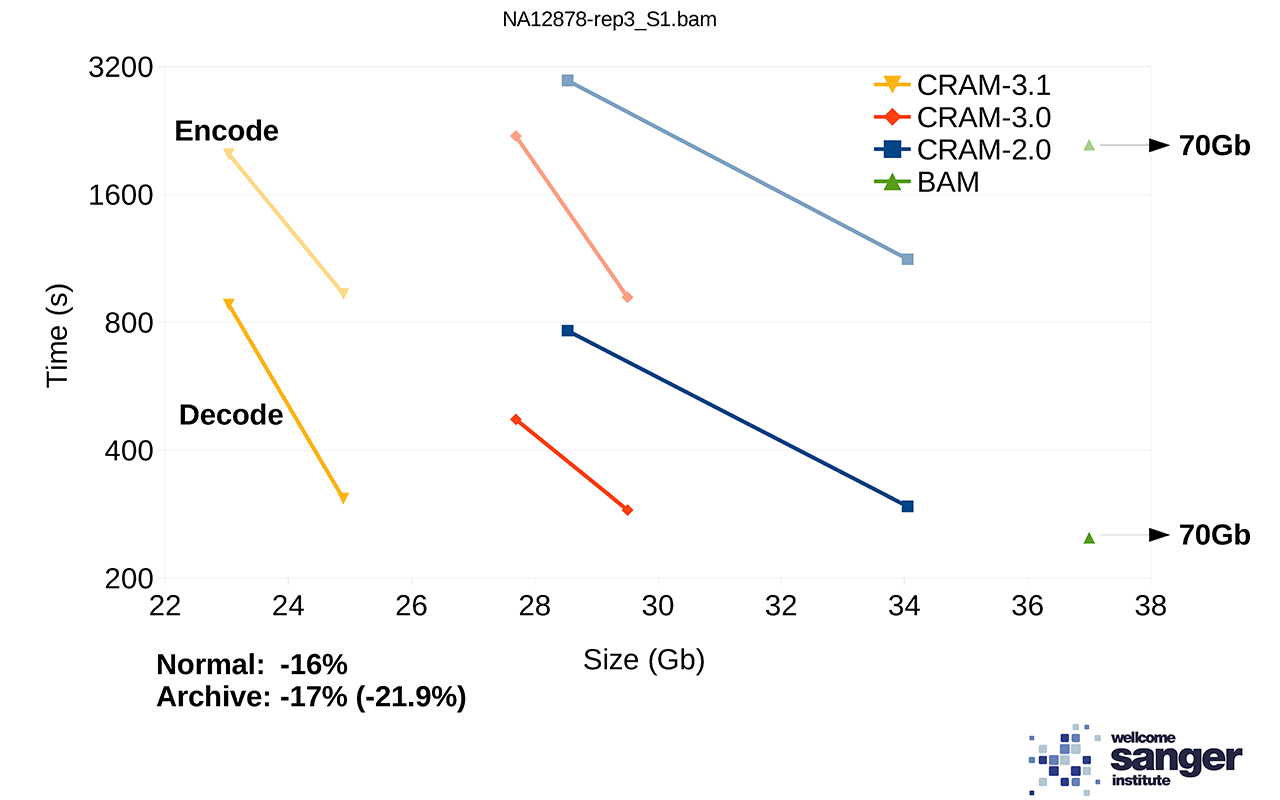 Figure 2: File sizes and times for encoding and decoding 1 million NovaSeq alignments, each line showing two points in the trade-off between size vs time.
Figure 2: File sizes and times for encoding and decoding 1 million NovaSeq alignments, each line showing two points in the trade-off between size vs time.
Ideally we don’t wish to waste time decoding bits of data that we are not interested in. Selecting specific data types (e.g., SAM columns) within CRAM is very feasible. For example, try using Samtools flagstat on a BAM vs a CRAM file and you’ll see it’s much faster in CRAM, as it has selectively skipped decoding of some fields. Similarly, indexing a CRAM is an order of magnitude faster than BAM.
Technically Speaking
The CRAM format gives complete freedom to write data in whichever blocks it likes so if the encoder puts all data in one block, there is no way to select only some types. However, the major CRAM implementations are now putting each type of data into their own blocks to facilitate ease of decoding (and have been doing this for a while). This means we can get certain types of data out much faster.
In some cases it’s possible to do the same for writing too. For example, if we wish to drop one single tag, say the large OQ field, then we don’t want to decode sequence, name, quality, and more only to immediately reencode them again. We’d be better off simply copying the compressed data over, bar the block containing the OQ field and the meta-data that indicates its presence. This isn’t possible within Samtools / htslib, but the cram_filter tool demonstrates some of this in the -t option which permits dropping of specific auxiliary tags. It can do this with little more than a read/write loop, which—even with the effort to adjust the tag list and meta-data blocks—comes out at least 50 times faster than a traditional transcoding loop:

If we can do it with removal, in theory we can do it with addition too, but this exposes an issue with our software implementations: again this isn’t a CRAM file format woe, but a programming problem.
Say we wish to add an auxiliary tag stating the average quality value, we’d only need to decode the quality from the CRAM file. We know what to do—decode the quality block, add a new tag, update various CRAM meta-data blocks, and do read/write loops on all the other blocks. (Of course this also doesn’t work too well if we want to strip out entire reads, but we could simply mark them as unmapped and leave in-situ.)
Htslib doesn’t currently have a good API for block level manipulations, but this is an implementation detail and not an issue with the file format itself.
Finally, note that it is also possible to extract regions of CRAMs with zero transcoding using cram_filter (again using the -n option I mentioned before, although it could really do with a saner interface that permits e.g. X:10000000-20000000). This, along with BAM equivalents, are the backbone of the efficient GA4GH htsget protocol for streaming NGS data.
This is a peculiar one, but I’ve seen it written that CRAM suffers as it leaves no room for efficient implementations, due to dictating the encoding process instead of the decoding process.
That is simply not true.
The CRAM format specifies the file layout and the decoding method, but the encoder has complete freedom on how to break the data up.
Technically Speaking
Encoders can decide which data types to put in which blocks and with which codec. Furthermore, CRAM permits storing multiple data series in the same block if there is strong correlation between values and a mixture would lead to better compression ratios. In the early days we gradually learned how to better encode CRAM 2.0 files which led to Scramble producing files smaller than Cramtools of the same era. (Cramtools implemented the same ideas, so it’s now comparable again.)
Is it perfect? By no means! But that is what open standards development is all about—come to the file formats task team meetings and help us ensure the next version of CRAM suits your needs.


